| 17 ALFA HIDROXIPROGESTERONA
Método: RIA, HPLC
Muestra: suero o plasma. Estable a 4°C por 4 días
o a –20°C por períodos mayores de tiempo. En la mujer
se recomienda realizar la determinación entre los días 3
y 5 del ciclo por la mañana.
Screening neonatal: sangre en papel de filtro. (Ver
Actualización: Screening Neonatal: Hiperplasia Suprarrenal Congénita).
Valor de referencia:
RIA:
Femenino:
Fase folicular temprana: 0,2-2,6 ng/ml
Fase lútea: 0,3-4,0 ng/ml
Post menopausia: 0,1-1,2 ng/ml
Masculino: 0,4-3,5 ng/ml
Significado clínico:
La 17  hidroxiprogesterona es el sustrato
para la hidroxilación en posición 21 en la biosíntesis
del cortisol. hidroxiprogesterona es el sustrato
para la hidroxilación en posición 21 en la biosíntesis
del cortisol.
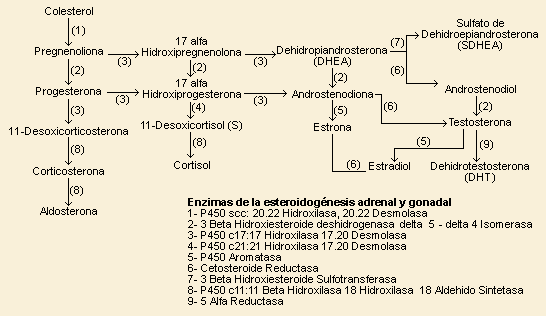
Las dos enzimas críticas en la biosíntesis de cortisol
son la 21 hidroxilasa y la 11-ßhidroxilasa. La 21 hidroxilasa es
la enzima responsable de la conversión de progesterona a deoxicorticosterona
(DOC) en la zona glomerulosa y de 17-hidroxiprogesterona
a deoxicortisol en la zona fasciculada. En la deficiencia de esta enzima
se acumula su precursor principal: 17-hidroxiprogesterona
en sangre.
La hiperplasia suprarrenal congénita (HSC) es un desorden heredado
en forma autosómica recesiva ocasionado por un defecto en una de
las 5 enzimas que intervienen en la esteroideogénesis adrenal.
Varios son los déficit enzimáticos descriptos en la estroideogénesis
suprarrenal siendo: el déficit de 21 hidroxilasa el más
frecuente (aproximadamente 95% de todas las formas de hiperplasia suprarrenal
congénita), seguido por el déficit de 11-ßhidroxilasa
(5-8%), el de 3-ßhidroxiesteroidehidrogenasa y el déficit
de 17 hidroxilasa.
Esto trae aparejado una disminución en la producción de
cortisol y en algunos casos también de aldosterona.
La incidencia de HAC clásica debido a deficiencia de 21 hidroxilasa
basada en estudios restrospectivos ha sido reportada por muchos investigadores
desde 1950 y ha variado desde 1/490 a 1/67000 nacidos vivos.
La incidencia en nuestro país es de aproximadamente 1/2503. .
La prevalencia de HSC es elevada entre la población judio-ashkenazies,
en la población general es de 1/100 y baja en la población
japonesa es de 1/20000.
La prevalencia de HSC es elevada entre la población judio-ashkenazies,
en la población general es de 1/100 y en la población japonesa
es de 1/20000.
Debido al patrón de herencia (autosómico recesivo) deben
estar ambos alelos afectados para que se desarrolle la enfermedad.
Las mujeres pueden presentar genitales ambiguos debido a la exposición
prenatal de elevados niveles de andrógenos; virilización
post-natal o crisis addisoniana perdedora de sal neonatal. Los varones
presentan pubertad precoz durante la infancia y crisis perdedora de sal
neonatal. Ambos sexos manifiestan post natalmente crecimiento somático
rápido con una acelerada maduración esquelética,
cierre temprano de epífisis y corta estatura adulta. Otros síntomas
de exceso de andrógenos incluyen patrón anormal de vello
corporal y disminuida fertilidad debido a una disrupción del eje
hipotálamo-hipofiso-gonadal.
El gen que codifica para la deficiencia de 21 hidroxilasa se encuentra
ubicado en el brazo corto del cromosoma 6, muy próximo al lugar
en el cual codifica el antígeno mayor de histocompatibilidad. En
estudios realizados a pacientes con parientes previamente afectados se
estableció que toda la descendencia afectada es HLA idéntica.
Resultando ser el genotipo HLA marcador del genotipo de HSC.
Existen dos formas clínicas de presentación de la deficiencia
de 21-hidroxilasa:
- Deficiencia clásica, que involucra el bloqueo parcial o total
de la actividad enzimática y se presenta clínicamente
al nacimiento. Esta incluye la forma virilizante simple y la perdedora
de sal.
- Deficiencia no clásica o de comienzo tardío, que involucra
solamente un bloqueo parcial de la actividad enzimática.
También está la forma críptica sin manifestaciones
clínicas.
En la forma clásica los precursores de la 21-hidroxilasa se encuentran
marcadamente elevados mientras que en la forma no-clasica los valores
están moderadamente elevados o normales. Entre portadores y la
población general los valores de 17 -hidroxiprogesterona
pueden superponerse.
Del 1al 2 % de los hiperandrogenismos post-puberales se deben a HSC no
clásica. En los hombres el aumento de ACTH puede conducir a un
adenoma (40% de los casos), hiperinsulinismo.
Las consecuencias clínicas de la deficiencia de 21-hidroxilasa
derivan de la superproducción y acumulación de los precursores
próximos al paso enzimático bloqueado desviándose
hacia la síntesis de andrógenos.
La 17 hidroxiprogesterona puede ser medida en
sangre en papel de filtro para el diagnóstico prenatal de hiperplasia
suprarrenal congénita. (Ver
Actualización: Screening Neonatal: Hiperplasia Suprarrenal Congénita).
Utilidad clínica:
Diagnóstico de hiperplasia suprarrenal congénita de comienzo
tardío, debido a deficiencia de 21 hidroxilasa.
Valores de 17 a-hidroxiprogesterona inferiores a 2 ng/ml >> Descartan
la HSC (no clásica)
Entre 3,0 y 5,0 ng/ml >> Sospecha HSC no clásica. Realizar
prueba de estimulo con ACTH. (Ver prueba
de estímulo con ACTH - Prueba Rápida)
Mayores de 5,0 ng/ml >> Confirma el diagnóstico.
Diagnóstico diferencial de:
Síndrome de ovarios poliquísticos.
Hiperandrogenismo suprarrenal sin defecto de la enzima.
Tumores virilizantes ováricos o suprarrenales.
Obesidad central con resistencia a la insulina.
Síndrome de Cushing.
Hirsutismo idiopático o por aumento de 5 a reductasa.
Monitoreo del tratamiento en pacientes con HSC.
Evaluar pacientes con hiperandrogenismo.
Screening neonatal. (Ver
Actualización: Screening Neonatal: Hiperplasia suprarrenal congénita).
Variables preanalíticas:
Aumentado:
La 17 hidroxiprogesterona
tiene ritmo circadiano marcado con valores mayores durante la mañana.
Las elevaciones encontradas en la fase lútea indican la actividad
del cuerpo lúteo.
Embarazo, prematuros.
Variable por enfermedad:
Aumentado:
Neoplasias adrenales u ováricas, hiperfunción adrenocortical
(exceso de mineralocorticoides), deficiencia de 11 ß-hidroxilasa asociado
a modestas elevaciones.
Disminuido:
Insuficiencia adrenal (enfermedad de Addison), pseudohermafroditismo.
Variables por drogas:
Disminuido:
Glucocorticoides, testosterona.
Bibliografía:
1. Tietz N. W. Clinical Guide to Laboratory test, edited by W.B. Saunders
Company, third edition, United States of America ,1995.
2. Young D. Effects of Preanalytical Variables on Clinical Laboratory
Test. AACC, second edition, 1997.
3. Young D. and Friedman R. Effects of Disease on Clinical Laboratory
Test, edited by AACC, third edition, 1997.
4. Burtis C. and Ashwood E. Tietz Textbook of Clinical Chemestry, W.B.
Saunders Company, of America,1999.
5. LotharT. Clinical Laboratory Diagnostics: Use and assessment of clinical
laboratory results, English edition, 1998.
6. Guitelman, Aspiz. Exploración funcional endócrina. Editorial
Akadia. Buenos Aires, Argentina, 1992.
7. Wilson & Foster. Textbook of Endocrinology. 8 th Edition W.B. Saunders
Company 1992.
8. Jacobs D.S., Demott W.R. Grady H. et al. Laboratory Test Handbook,
Edit by Lexi-Comp Inc., Cleveland, United States of America, 4th edition,
1996.
9. Ricardo Azziz, Didier Dewilly and David Owerbach. Clinical Review.
Review 56. Nonclassic Adrenal Hyperplasia. Current Concepts. J. Clin.
Endoc. Metab. Vol 78 Nº4, 1994: 810-815.
10. Talia Edgar Geva, MD, Arye Huwirtz MD and Ariel Rösler, MD. Secondary
biosynthetic defects in women with late-onset congenital adrenal hyperplasia.
New England Journal of Medicine, 855-863
|